Logistic regression and GWAS¶
约 1117 个字 预计阅读时间 4 分钟
全基因组关联研究(GWAS,Genome-Wide Association Study),是一种病例-对照研究设计。研究选取很多人类个体,并分为患病组和未患病组。通过使用机器学习方法来比较两组个体的基因组数据,研究者可以找出可能与疾病相关的基因变异。GWAS 的目标是找到与疾病相关的常见基因变异,以此帮助理解疾病的遗传基础。
首先,研究者收集个体的基因序列信息。我们重点关注于单核苷酸多态性(Single nucleotide polymorphism, SNP),即DNA序列中一个碱基对的变化对疾病表现型的影响。例如父母的某个位点上可能有一个碱基为C,但子代中可能会有一个变为T的变异位点。这个变异位点正是GWAS研究的重点。
由于每个个体都有23对染色体,因而所有受试者在 DNA 上有配对的等位基因,序列采集后会得到两组基本相同的序列(可能含有等位基因变异)。我们因此可以得到碱基变异的剂量(Dosage Information)。
-
0剂量:个体的两条染色体上都是参考等位基因(0/0)。
-
1剂量:一条染色体是参考等位基因,另一条是备选等位基因(0/1)。
-
2剂量:两条染色体上都是备选等位基因(1/1)。
在实际研究中,基因变异的数据以VCF格式存储。VCF文件中包含了:
-
染色体和位置信息(Chromosome & Position)
-
参考等位基因(Ref)和备选等位基因(Alt)
-
每个个体的基因型信息(Genotype),如0/1、1/1等,表示该位点上参考和备选等位基因的组合。
-
该变异的注册ID编号(ID)。
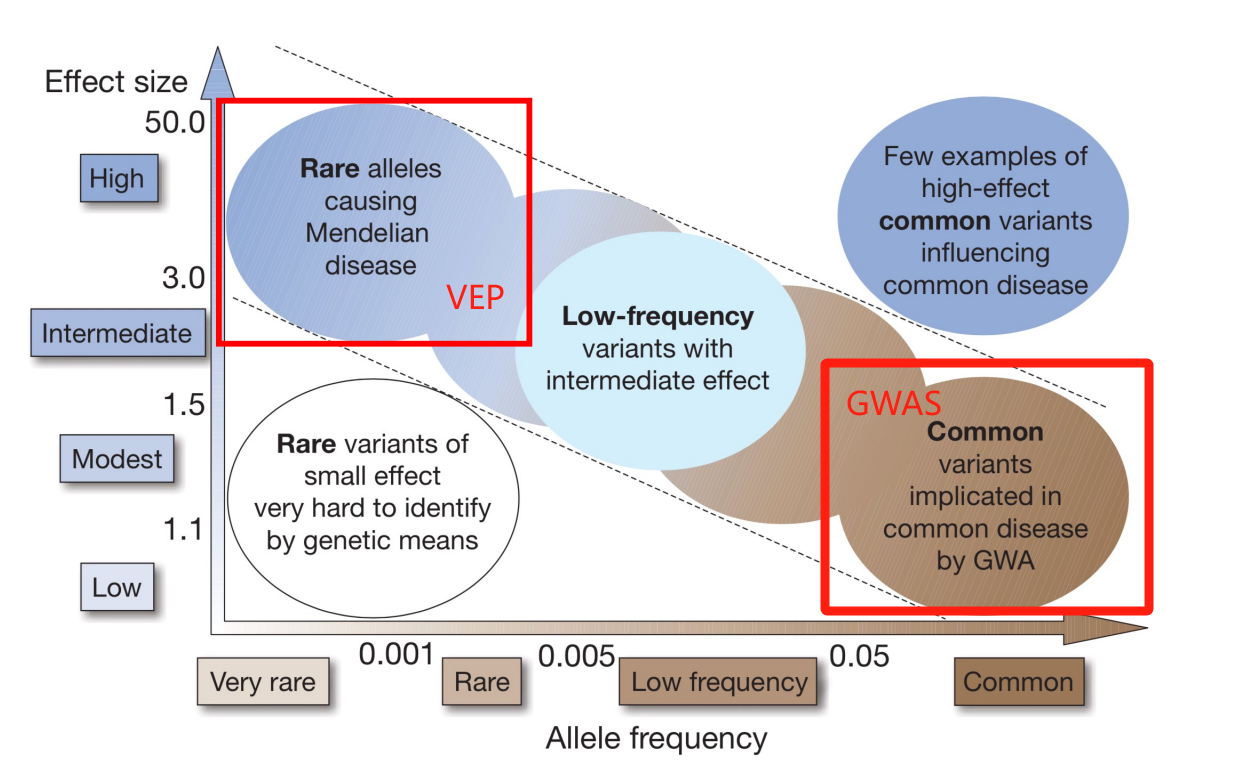
Logistic Regression¶
接下来我们明确我们的任务目标。我们希望通过分析每个 SNP 与疾病发生的相关性,最终通过统计检验获得与疾病显著相关的 SNP。
在 GWAS 中,我们希望使用 Logistic 回归,来得到每一个变异剂量 \(X\) \((X=0,1,2)\) 对应的疾病的可能性 \(P(Y=1)\)。
Logistic 回归的目的是通过回归系数 \(\beta\) 来估计 SNP 剂量 \(X\) 与 疾病状态 \(Y\) 之间的关系。回归模型如下:
其中:
-
logit 函数:定义为 \(\text{logit}(p) = \ln \left( \frac{p}{1-p} \right)\),是为了将概率 \(p\) 映射到实数范围来对应 \(\beta_0+\beta_1 X\)。
-
\(P(Y=1)\) 是个体患病的概率,即 \(Y = 1\) 的概率。
-
\(\beta_0\) 是截距。
-
\(\beta_1\) 是自变量 X 的回归系数。
我们希望通过已知的 X 和 Y 来估计 \(\beta_0\) 和 \(\beta_1\)。
解出这个方程,我们可以得到:
这就是 Logistic 回归的核心公式,通过这个公式我们可以预测某个个体的患病概率 \(P(Y=1)\),其输入是 SNP 剂量信息 X。
为了得到 \(\beta_0\) 和 \(\beta_1\),我们使用极大似然估计法(Maximum Likelihood Estimation, MLE)。MLE 的目标是找到让整体观测数据 \((X, Y)\) 的可能性最大的 \(\beta_0\) 和 \(\beta_1\)。
假设有 \(n\) 个个体的数据。对于每个个体 \(i\),观测值 \(Y_i\) 是 0 或 1,概率为 \(P(Y=1|X)\),即:
这个似然函数表示的是给定每个 \(X_i\) 值时,\(Y_i = 1\) 或 \(Y_i = 0\) 的概率。
为了简化计算,我们取对数得到对数似然函数:
通过最大化对数似然函数,求解出 \(\beta_0\) 和 \(\beta_1\)。
我们接下来使用梯度下降法迭代求解最佳的 \(\beta_0\) 和 \(\beta_1\)。
其中,\(\alpha\) 是学习率。
反复迭代,直到对数似然函数收敛,得到最优的 \(\beta_0\) 和 \(\beta_1\)。
一旦通过梯度下降法或其他优化算法求得了 \(\beta_0\) 和 \(\beta_1\),我们就可以对模型进行解释:
-
\(\beta_1\) 是 SNP 剂量 \(X\) 对患病概率 \(P(Y=1)\) 的影响。如果 \(\beta_1 > 0\),表示 SNP 剂量越高,患病的概率越高;如果 \(\beta_1 < 0\),表示 SNP 剂量越高,患病的概率越低。
-
使用估计得到的 \(\beta_0\) 和 \(\beta_1\) 可以计算每个个体的患病概率:
statistic analysis¶
通过上面的方法,我们得到了每一个 SNP 变异位点对应的疾病发生的概率。例如,当某个体出现第 \(i\) 个 SNP 变异位点,并通过这个模型计算出该个体在 SNP 位点上的剂量值 \(X_i\) 所对应的概率 \(P(Y=1∣X=i)=0.9\),那么我们可以人为该个体有 90% 的概率患病。
然而,由于 GWAS 通常会对大量的 SNP 进行分析(通常上百万个),这就引入了一个问题,即我们可能会得到很多假阳性(false positives),这意味着我们错误地认为某些 SNP 与疾病有关,而实际上它们并没有关系。
因此我们需要来进行统计学分析,尝试降低这部分产生的假阳性,提高准确度。
在 GWAS 中,我们对每个 SNP 都会进行一个假设检验,假设原假设为 \(H_0: \beta_1 = 0\)(即该 SNP 对患病没有影响)。通过 Logistic 回归分析得到的回归系数 \(\hat{\beta}_1\) 和标准误差 \(SE(\hat{\beta}_1)\),可以计算出 Z 值:
Z 值可以用来计算 P 值,它表示如果原假设为真(即该 SNP 对疾病没有影响),我们会观察到如此极端的检验统计量的概率。P 值越小,说明该 SNP 与疾病的关联越强。
2. 显著性阈值 \(\alpha\):¶
为了判断一个 SNP 是否与疾病显著相关,通常会设定一个显著性水平(例如 \(\alpha = 0.05\)),这表示我们接受最多有 5% 的假阳性率。如果 P 值小于 \(\alpha\),我们就可以拒绝原假设,认为该 SNP 与疾病有关。
然而,当我们在 GWAS 中分析大量的 SNP 时,这样的显著性水平会导致很多假阳性。假设我们测试了 1,000,000 个 SNP,如果每个 SNP 的显著性水平为 \(\alpha = 0.05\),那么理论上会有 5% 的假阳性,也就是:
这就意味着在 1 百万个变异位点中,我们可能会得到 50,000 个假阳性,即错误地认为 50,000 个 SNP 与疾病相关,而实际上它们并没有关联。
3. 多重假设检验校正:¶
为了控制假阳性率,GWAS 采用了多重检验校正方法。图片中提到了两种常见的校正方法:
- Bonferroni 校正:
- 这是最常用的一种校正方法,用来控制至少一个假阳性的出现概率。
- 它通过将显著性水平 \(\alpha\) 进行调整,除以测试的总数目 \(m\),即新的显著性水平为:
例如,如果我们测试 1,000,000 个 SNP,并设定 $\alpha = 0.05$,那么 Bonferroni 校正后的显著性水平为:
只有 P 值小于这个非常小的阈值时,我们才会认为该 SNP 与疾病有显著关联。
- Benjamini-Hochberg 校正(FDR 控制):
这种方法控制的是错误发现率(False Discovery Rate,FDR),即我们发现的显著结果中有多少比例是假的。它是一种更宽松的校正方法,允许一定比例的假阳性,从而增加检测显著 SNP 的能力。FDR 控制的方法在 GWAS 中也广泛使用,尤其当我们不需要严格控制每一个 SNP 的假阳性,而是希望在假阳性率可控的情况下找到更多的关联位点时。
为什么我们更多的不希望有太多的假阳性?¶
假阳性结果会误导研究者,使他们去进一步研究那些实际上与疾病没有关联的 SNP,浪费时间和资源。
妈的,概率论没学好,先弃坑了。